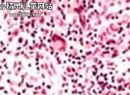
小儿遗传性球形红细胞增多症(5)
(2)血影蛋白与锚蛋白的结合部分缺乏:血影蛋白与锚蛋白结合部分缺乏的生物化学表现最先在1988年由Coetzer等提出。锚蛋白代表血影蛋白在膜上的主要连接部位,因此,尽管血影蛋白的合成正常,锚蛋白缺乏常伴有相应比例的血影蛋白减少是不奇怪的。例如,β-血影蛋白突变的HS,大多数的锚蛋白缺陷属于与mRNA累积减少相关的点突变。
除外锚蛋白Florisnopolis外,移码突变与严重的HS有关,这一点由来自3个不同遗传背景家族的HS患者而证实。现有的报道中15%~20%的锚蛋白基因(ANKl)突变属新生突变(de novo mutation)。在两个家庭中发现有双亲镶嵌体锚蛋白突变,因此,在相同HS家族中有临床症状不同的病例发生。包括锚蛋白基因部位的缺失或移位的染色体组型异常的非典型HS病例也有报道。1例患者,8号染色体上全部锚蛋白基因缺失导致一个大的缝隙缺失。锚蛋白缺失可能是典型症状的球形红细胞增多、智力低下、典型面容和性腺功能减低的邻近基因综合征的一部分。
(3)带3蛋白部分缺乏:带3蛋白部分缺乏发现于轻、中型显性遗传的HS患者,伴有蘑菇样或钳形红细胞,其中大部分患者伴有蛋白4.2缺乏。迄今为止,发现有近50种不同的带3突变与HS有关,这些突变延伸遍及带3,存在于胞浆区域和膜剪切区域。带3突变的纯合子(带3Coimbra)可引起致死性或近致死性的HS,伴有胎儿水肿、代谢性酸中毒以及与带3完全缺失并蛋白4.2缺乏相关的严重贫血。影响带3蛋白的等位基因(SLC4A1)已被鉴定,当带3蛋白突变连续遗传,将加重带3缺乏并使疾病的临床症状更加恶化。
某些带3缺乏的HS病例,网织红细胞减少。这种病例带3合成和mRNA水平是正常的。同时也发现某些病例存在带3蛋白不稳定。引起带3缺失的机制尚不清楚,推测机制有:减弱带3蛋白与锚蛋白的连接,导致初始带3蛋白或锚蛋白缺乏;或膜上缺少带3组合的“零件”。在某些带3缺乏的HS患者,带3基因表达可以降低,或一个带3突变可妨碍带3进入内质网膜的固有联合转录插入,或妨碍带3移位到原生质膜。有些带3突变集簇于膜的山梨醇脂肪酸酯(跨度)区域,这些膜的山梨醇脂肪酸酯(跨度)区域替代了大范围的保守精氨酸,这些被替代的精氨酸全部定位于细胞质的跨膜螺旋的末端,维持跨膜山梨醇脂肪酸酯片断的方向。