С��������-6-��������øȱ��֢��ʲôԭ������
����������ԭ��
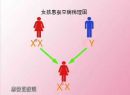
������֢��һ����������ȫ�����Ŵ�������ͻ�����λ�� X Ⱦɫ���ϣ�������ԣ��Ӻ���Ů�� G6PD ����ƫ�ͣ�������Ѫ��������Ů�Կɷ����������ټ������� G6PD �Ļ���ʸ��ӵĶ�̬�ԣ����γɶ��� G6PD ȱ��֢�ı����͡��ò������У��϶�������ҩ�������ʹҩ���ǰ�ҩ��������ࡢ����ୡ�ά����K����ˮ����ȣ���Ⱦ����ԭ����ϸ������
�����Ŵ�ѧ�о���1986�꣬Persico��Martlni�ȷֱ��ò�ͬ�ķ����ɹ��ؿ�¡����G-6-PD���������cDNA���У��Ӷ�ʹG-6-PD���о����뵽����ˮƽ��ʹ�����ܴӻ���ˮƽȥ̽��G-6-PDȱ���ĵ�����һ���ṹ�ı䡣1991��Ellson�Ȳⶨ����G-6-PD������ȫ˳�� G-6-PD����Լ18kb����13�������Ӻ�12���ں�����ɣ�����һ����515�����������G-6-PD�����ʡ���������Ӧ�ÿ�¡G-6-PD��������PCR����ֱ�����з����Ѽ�����120�����Ŵ�ѧ�����ͣ����г�3��Ϊ������ȱʧ�⣬���Ϊ������������û���G-6-PD������һ�����һ���(homekeeping gene)����˶���������DZ���ģ�����G-6-PD������ȫɥʧ��ͻ��(��ȱʧ������ͻ��)�����������ģ���������1��3��13�ⶼ�ѷ��ֵ�ͻ�䡣�й������ѷ���15�ֵ�ͻ�䣬�����о�֤ʵ��ͬ����ͬ���廼����50%����Ϊ1376G→T��1388G→A�����������ϸ����Ѫ��ƶѪ��ͻ�伯�е���ø���ǻ�ĩ�ˣ���362��446���������Ƭ�Σ����ֵ�������������ͻ��������ø�İ���ĩ�ˡ������˸���Ȥ����G-6-PD A-��ͻ�䡣A-�����Ŵ������ԣ�����2����λ�����˼���û�������һ����376A→G����һ��������202G→A��680G→A��968T→C��A-�����������е�Ƶ��Ϊ12%����һ���ڷ������г����ı�����ΪG-6-PD A�������������е�Ƶ��Ϊ20%��G-6-PD A��ͻ��Ϊ376A→G������G-6-PD A-��һ���е�һ��ͻ�䡣��ˣ�Beutler����ΪG-6-PD A-�����Ǵ�G-6-PDB(Ұ����)→G-6-PD A→G-6-PDA-��������Ȼѡ��(����ű��)A-�ĸ�Ƶ�ʵ��Ա�������������ͳ�����������Ϊͬһ��G-6-PD�����������п����Dz�ͬ�Ļ���ͻ�����¼���ʵ���Dz�ͬ�Ļ�������͡���G-6-PD(-)��3�����͵Ļ���ͻ�䣺��202G→A��376A→G;��680G→T��376A→G;��968T→G��376A→G����ǰ��Ϊ��һЩ�Dz�ͬ�����������ͣ���ʵ����ͬһ���ͻ�����¡���G-6-PD����������Kaiping��Anant��Dhon��Petrieh-like��Sappoto-like��Ϊ1388G→Aͻ��(463 Arg→His)�� G-6-PD����λ��X q28��G-6-PDȱ��Ϊ�������IJ���ȫ�����Ŵ�����ˣ����б����������Իᷢ����Ů��G-6-PDȱ���Ӻ�����������ϸ��Ⱥ��G-6-PDȱ��ϸ��������ϸ����G-6-PDȱ��ϸ��������ϸ���ı����仯�ܴ�һЩ�Ӻ���Ů�Ա���Ϊ��ȫ��������һЩ�����Ϊ��ȫ�쳣��G-6-PD�Ӻ��ӱ��ֵ�����������������XȾɫ��ʧ����̵�ijЩ���ԵĽ������ΪXȾɫ��ʧ��������ģ���ʱ����ĸ���XȾɫ���ǻ��ϸ����¡����ֳ���ơ���XȾɫ��ʧ��ͳ����ڼ侭�������ϸ������ʹijһ�ֿ�¡����һ��ֻ�к�С��ѡ���������ƾͻᵼ��������ȱʧϸ����֮�������IJ��죬�������Ů���Ӻ�������Ѫ��G-6-PDȱʧ��ϸ����������ϸ��֮�ȵ����������Բ���ͻᵼ�����ٴ����ָ��졣
| ��������� | �� �̶�С����ʲô���ã� | �� ��������һ��Ҫ���÷��;� |
| �� ��������һ��Ҫ���÷��;� | �� ����϶����кð취�� |